|
 HEMOPHILIA: LIVING,
AND BEING SAFELY ACTIVE, WITH A BLEEDING
DISORDER HEMOPHILIA: LIVING,
AND BEING SAFELY ACTIVE, WITH A BLEEDING
DISORDER
Written by: Brittany
Maillet, University of Florida
Hemophilia is
a bleeding disorder which is largely
inherited, and affects 18,000 people
in the United States (1).
The disease is the result of a missing
or deficient blood clotting protein
and there are two types, including hemophilia
A, which involves the factor VIII protein,
and hemophilia B, which involves factor
IX (2).
Hemophilia A is much more common, and
affects 1 in every 5,000 males born
in the US while hemophilia B only affects
1 in every 25,000 males born (2).
Hemophilia is
inherited through the X chromosome;
therefore women are generally only carriers
of the disease and men actually have
it. A woman who is a carrier of hemophilia
has a 25% chance of giving birth to
a baby girl who is a carrier of the
disease, as well as a 25% chance of
having a boy who actually has hemophilia.
If a man with hemophilia has a boy,
there is no chance of the boy inheriting
the disease, however if he has a girl
she will be a carrier no matter what
(2).
Hemophilia can
range from mild to severe, and can lead
to spontaneous internal bleeding and
bleeding following injuries or surgery
(1). Mild
hemophilia may only be detected in adulthood
after some sort of trauma or surgery
has occurred, while moderate hemophilia,
affecting 15% of the population of people
with hemophilia, may cause bleeding
after injuries as well as spontaneous
bleeding episodes (2).
The severe form of the disease affects
about 60% of people with hemophilia,
however, and involves frequent spontaneous
bleeding episodes which often occur
in the joints and muscles (2).
This leads to
the most frequent and severe manifestation
of hemophilia: hemarthrosis,
which occurs due to repeated joint bleeds
and is often initiated at a young age
(3). Hemarthrosis
occurs when “blood within a joint
results in inflammation and hypertrophy
of synovial
membranes, causing increased vascularization
of the joint and bone degeneration”
(3, p1).
It is also associated with pain and
decreased mobility, and prevention through
aggressive treatment methods which will
be discussed later in the paper ensure
the best outcome for pediatric patients
dealing with hemophilia.
A common misconception
associated with the disease is that
a small cut will result in perfuse and
uncontrollable bleeding, but this is
not the case. A person with hemophilia
does not bleed any heavier or faster
than someone without the disease; the
only difference is that they bleed longer.
The normal coagulation progression controls
bleeding through a complex process involving
20 different clotting factors (2).
The sequence of events our bodies go
through to stop a wound from bleeding
begins when vessels constrict and platelets
pile up around the cut.
Next, a clot
is formed as calcium, proteins, platelets,
and other tissues react together and
strengthen with time, eventually forming
a substance called fibrin
which stops the bleeding (2).
In the case of a person with hemophilia,
however, one of the clotting factors
is not present or does not work the
way it should, preventing the fibrin
from effectively stopping the bleeding.
Common bleeds associated with people
who have hemophilia include bruising,
joint bleeds, nose bleeds, mouth bleeds,
and deep muscle bleeds (2).
Serious life threatening bleeds can
also occur, however, and sites to pay
special attention to include the eyes,
head, neck, abdominal region, and kidney/bladder
area (2).
Research on
hemophilia has made monumental strides
in the past couple of decades, resulting
in a much better quality of life for
those living with the disease. Until
the 20th century, the cause of excessive
bleeding was unknown and relatively
uninvestigated. In the 1930’s,
platelet-free plasma was discovered
to help the clotting problem, and in
the 40’s it was discovered that
two different forms of the disease occurred
based on which protein factor was missing
or deficient (2).
Treatment was
still limited to icing joints and entire
blood transfusions, and life expectancy
hovered at around 30 years of age(2).
Many men died in early childhood, most
commonly of brain bleeds, and hemophilia
existed as one of the most painful diseases
known because of hemarthrosis.
The 1950’s and 60’s brought
a new knowledge of the coagulation process,
and cryoprecipitate
was also discovered. Cryoprecipitate
is the precipitate that is left after
thawing plasma, and it is rich in factor
VIII (2).
This was a major breakthrough because
it replaced high volume plasma transfusions
and could be infused to control serious
bleeding.
In the 1970’s
cryoprecipitate was taken to the next
step and made into freeze dried powder
which could be kept at home, allowing
people with hemophilia to self infuse
themselves in the case of a bleed and
eliminating the need for many hospital
visits (2).
The 1980’s brought an entirely
new struggle to the already difficult
forefront of the disease, however, as
HIV/AIDS emerged and infected half of
the people with hemophilia in the United
States. Not until the 1990’s was
a new and entirely safe method of retrieving
factor developed when recombinant technologies
allowed it to be manufactured (2).
Recombinant
factor is the result of the isolation
and cloning of cDNA from human factor
VIII, which paved the way for biosynthesis
of genetically engineered factor VIII
(4). This
recombinant factor is structurally and
functionally identical to that of human
plasma derived factor, and elicits the
same physiological response (4).
The difference, however, is that it
is much safer than plasma derived factor
since there is no danger of spreading
any blood-borne diseases such as HIV/AIDS.
Recombinant
factor is now mass produced and utilized
in prophylactic
therapy for children with hemophilia.
Prophylactic therapy involves an intense
preventive treatment regime with infusions
of factor relative to the child’s
body weight given on average every other
day. Many studies have been done comparing
the benefits of prophylaxis
to that of episodic therapy, which involves
injection of factor into the hemarthrosis
after it occurs rather than preventively.
Results show that prophylactic therapy
prevents chronic bleeding episodes,
allowing children to look forward to
a life with less pain and more active
lifestyles (2).
One recent study even showed that prophylaxis
with recombinant factor VIII was successful
in preventing hemarthrosis and joint
damage in 93% of participants, a much
better outcome than those who received
episodic treatment (5).
 Despite
prophylactic therapy’s undeniable
success in treating children with hemophilia,
the CDC reported that only 51.5% of
children under the age of 6 received
treatment with it in 2004 (5).
The number one reason for this is the
extremely high cost of recombinant factor
VIII, which can add up to more than
$300,000 per year depending on the size
of the child (5).
Other reasons include the amount of
time required for infusions, difficulty
in getting the child to cooperate, and
limited venous access (5). Despite
prophylactic therapy’s undeniable
success in treating children with hemophilia,
the CDC reported that only 51.5% of
children under the age of 6 received
treatment with it in 2004 (5).
The number one reason for this is the
extremely high cost of recombinant factor
VIII, which can add up to more than
$300,000 per year depending on the size
of the child (5).
Other reasons include the amount of
time required for infusions, difficulty
in getting the child to cooperate, and
limited venous access (5).
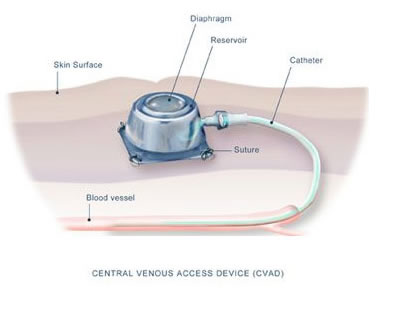
Prophylactic
treatment in young boys is generally
given through a fully implantable central
venous access device, commonly known
as a port. “A port consists of
a subcutaneous reservoir with a self
sealing silicone septum that is coupled
to a radiopaque silicone catheter (6).”
It is implanted into the upper chest
wall with the catheter fed into the
superior
vena cava, and a needle is used
to infuse the factor (6).
External central venous access devices
are also used sometimes, but ports are
preferable because they result in a
much lower infection rate and do not
require any daily cleansing or covering
(6).
Ports also present
no risk of being displaced during play,
which means the children can lead active
lifestyles. It does involve piercing
of the skin with a needle, however,
which can result in discomfort, but
when the port is implanted at a young
age children often become used to the
process. Ports should be left in only
for as long as clinically needed because
of the child’s limited venous
access, because this limits the risk
of complications such as thrombosis
(6).
It is important
to begin treatment of hemophilia at
the youngest age possible to avoid hemarthrosis
in the joints, because each successive
bleed brings the person closer to needing
a joint replacement. Without replacement
therapy and prophylaxis,
it is estimated that by the age of 20
a boy living with hemophilia will already
have five damaged joints, and most commonly
the knees, elbows, and ankles (7).
Although we
just discussed prophylaxis as being
the most effective preventive measure
for people with hemophilia, it is also
true that 80% of those living with hemophilia
reside in developing countries where
such an expensive treatment may not
be a feasible option (7).
In this case, pain and permanent damage
to the musculoskeletal system cause
restrictions in daily activity, and
physiotherapy and rehabilitation should
be used to prevent disabilities and
enhance autonomy and participation.
Rehabilitation is a multidisciplinary
approach including physiotherapy, occupational
therapy, and technical aspects, such
as orthoses,
which tries to reduce the impact of
disabling and handicapped conditions
and aid in achieving social integration
(7). Community
based rehabilitation and education about
living with hemophilia are crucial to
the process in cases where hemophilia
centers are not available.
If enough clotting
factor is not available to resolve bleeding,
physiotherapeutic methods should be
instituted next and may include rest,
cold, and compression. Active muscle
strengthening exercises should be done
whether the joint is postbleeding or
arthropathic,
which for a knee problem would mean
strengthening and stretching of quadriceps,
hamstrings, hip abductors and extensors,
and the calf muscles (7).
Exercise programs and even reverse dynamic
slings may need to be used to regain
range of motion in joints as well.
Pain needs to
be managed whether it be through the
use of oral medication, cold or warm
application, or other techniques specific
to the culture. Functional training,
as well as training of proprioception
and coordination is also crucial to
ensure restoration to normal activities
(7). This
includes integration of an individually
tailored program to help the patient
with daily living activities specific
to his lifestyle, whether that means
knee flexion for walking up and down
stairs or ankle flexion for squatting
on the floor. Proprioception
exercises should be gradually brought
in after treatment of acute hemarthrosis
has occurred, and some examples of these
exercises include balancing on one leg
with the knee slightly bent or running
in a figure eight (8).
Lastly, orthotics and shoe adaptations
are essential in treating musculoskeletal
problems, and can be created fairly
easily with local materials by technicians.
Thanks to factor
replacement and therapy, boys with hemophilia
no longer need to be overprotected and
sheltered from sports and other activities
typical of their age groups. In fact,
it is now recognized that sport and
exercise can reduce or prevent
intraarticular hemorrhages (9).
As with so many other disabilities,
it was first acknowledged that swimming
and hydrotherapy are the safest conditions
under which a boy with hemophilia can
participate in exercise. This type of
activity is easy on the joints and allows
for a wide range of motion without the
threat of causing any bleeds. Eventually
other activities were added to the list
such as bicycling, tennis, and hiking,
but many age appropriate activities
were still limited.
 As
more research was carried out and the
psychological impact of the disease
was studied, it became clear that participation
in sport could improve boys’ confidence
and self image and lead to a more independent
lifestyle (9).
Sports like baseball, and even football
in some cases, have become more acceptable
to participate in, and some proponents
recognize that factor infusion before
playing such sports can help eliminate
the threat of bleeds along with the
child recognizing his own limitations
(9). In
every case, contact sports are discouraged
somewhat but the decision about what
to participate in depends on the individual
and may require some trial and error. As
more research was carried out and the
psychological impact of the disease
was studied, it became clear that participation
in sport could improve boys’ confidence
and self image and lead to a more independent
lifestyle (9).
Sports like baseball, and even football
in some cases, have become more acceptable
to participate in, and some proponents
recognize that factor infusion before
playing such sports can help eliminate
the threat of bleeds along with the
child recognizing his own limitations
(9). In
every case, contact sports are discouraged
somewhat but the decision about what
to participate in depends on the individual
and may require some trial and error.
Author’s
Note:
Hemophilia is
of great interest to me because of the
prevalence of it in my family. My uncle
has the disease along with two of my
younger cousins, and I am a carrier.
After researching extensively the progression
of treatment for the disease, along
with its effects on the body, I can
clearly see the relation to my family
members. Since my uncle is around the
age of 40, he went through much more
as a child than my two younger cousins
because recombinant factor had not yet
been produced. My Grandma sometimes
talks about how often he had to be rushed
to the hospital for a blood transfusion
because of another bleed, and the constant
pain he was in because of the pressure
from hemorrhaging.
 Grandma
also remembers when freeze dried cryoprecipitate
became available and they stocked the
freezer at home with it. She said it
helped eliminate the need for some of
the emergency room visits because it
could get the internal bleeding under
control a lot of the time, but that
my Uncle Mark was still in a lot of
pain and extremely limited in physical
activity. Now that he is older he’s
already had multiple joint replacements,
and still endures physical pain every
day because of the hemarthrosis. I’ve
watched him many times infuse himself
with factor to control a bleed, and
some of his joints require it every
day now. Grandma
also remembers when freeze dried cryoprecipitate
became available and they stocked the
freezer at home with it. She said it
helped eliminate the need for some of
the emergency room visits because it
could get the internal bleeding under
control a lot of the time, but that
my Uncle Mark was still in a lot of
pain and extremely limited in physical
activity. Now that he is older he’s
already had multiple joint replacements,
and still endures physical pain every
day because of the hemarthrosis. I’ve
watched him many times infuse himself
with factor to control a bleed, and
some of his joints require it every
day now.
 In
comparison, my two little cousins are
seemingly completely unaffected by the
disease with the exception of infusion
time. They had ports put in before even
reaching the age of two, and my Aunt
infuses them with recombinant factor
VIII every other day. They are so used
to it that they just lie down on a pillow
and watch TV, hardly noticing the needle
prick or minding the 20 minutes it takes
out of their day. With this factor they
avoid bleeds and lead extremely active
lifestyles. In
comparison, my two little cousins are
seemingly completely unaffected by the
disease with the exception of infusion
time. They had ports put in before even
reaching the age of two, and my Aunt
infuses them with recombinant factor
VIII every other day. They are so used
to it that they just lie down on a pillow
and watch TV, hardly noticing the needle
prick or minding the 20 minutes it takes
out of their day. With this factor they
avoid bleeds and lead extremely active
lifestyles.
Both of them
play baseball, run around, and of course
wrestle because with four boys in the
house that’s hardly avoidable.
The only signs we see of the hemophilia
are a lot of bruising and pretty consistent
nosebleeds, but other than that the
prophylaxis
seems to control bleeds. In one case
Devin’s port became infected and
had to be changed immediately because
it caused an infection of the blood.
Neither boy reports joint pain, however,
nor do they let the disease slow them
down at all.
(another
great resource, and another...)
references
|

