Being Diagnosed with Amyotrophic Lateral Sclerosis
written by Melissa
Ortiz, University of Florida
This paper is dedicated to: the memory of Patricia Campbell
and her family
 Everyone
should be prescribed some exercise therapy, taking into account
a possible disability. Physical activity not only improves
health, but overall wellbeing. Careful examination of exercise
recommendations should especially be done for people who have
Amyotrophic
Lateral Sclerosis. Amyotrophic Lateral Sclerosis, also
known as ALS
or Lou Gehrig’s disease, is a motor neuron
disease that results from degradation of the motor nerve cells
(or motor neurons) in the brain and spinal cord, which then
evolves into progressive weakening of the muscles (Miller,
Gelinas, & O’Connor, 2004). Everyone
should be prescribed some exercise therapy, taking into account
a possible disability. Physical activity not only improves
health, but overall wellbeing. Careful examination of exercise
recommendations should especially be done for people who have
Amyotrophic
Lateral Sclerosis. Amyotrophic Lateral Sclerosis, also
known as ALS
or Lou Gehrig’s disease, is a motor neuron
disease that results from degradation of the motor nerve cells
(or motor neurons) in the brain and spinal cord, which then
evolves into progressive weakening of the muscles (Miller,
Gelinas, & O’Connor, 2004).
ALS has a late onset, a fast degradation,
and it leads to death; usually within 3 years of diagnosis.
The illness begins with fatigue, cramping, muscle weakness
and wasting of one or more limbs or fasciculation
of the tongue, before it gets worse (Miller, Gelinas, &
O’Connor, 2004). This is because the motor nerve cells
are the cells that transfer messages from the brain, telling
muscle what to do and when to do it. When the motor neurons
start dying, they harden and leave scar spots on the brain
or spinal cord. Scars make it harder for the brain to tell
muscles how and when to function, which eventually leads to
an inability to control those muscles.
Fasciculation of the tongue involves twitching and involuntary
contractions, caused by interference due to nerve death and
tissue hardening in signaling paths (motor neurons) from the
brain to the muscles. All eventually leads to a person losing
control of voluntary movement, and in some cases - breathing.
ALS is called Lou
Gehrig's disease after the world-renowned baseball player
for the New York Yankees who held a 60 year record for the
most consecutive baseball games played by any baseball player
(Miller, Gelinas, & O’Connor, 2004). He was one
of the best baseball players of all time. Until his quick
health degradation, Gehrig
was always extremely fit and popularly called "The Iron
Horse" for this durability, strength, and reliability
(Eig, 2005). He's still a sign of heroism in light of a destructive
condition.
As a person ages past 44 years old, the chance of being diagnosed
with ALS increases - reaching a peak at 75-84 years of age.
Then the chance of being diagnosed with ALS decreases. This
is demonstrated in figure 1 below (Majoor-Krakauer, Willems,
& Hofman, 2003). As can be seen, incidence rates are generally
higher in men than in women.
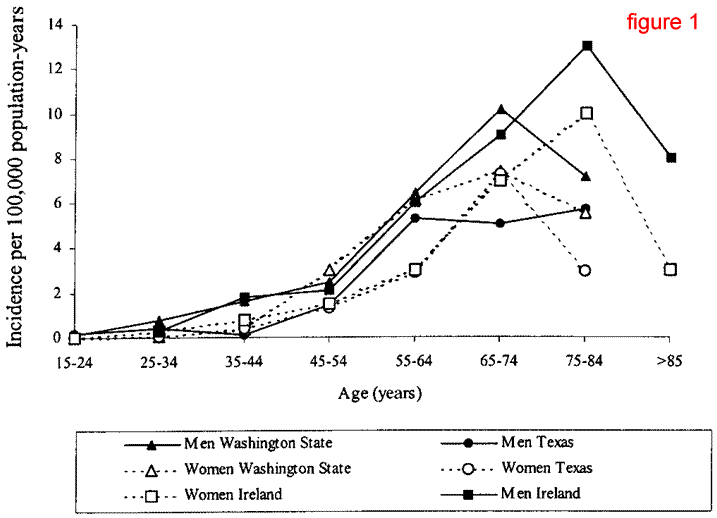
Figure 1 represents age-specific incidence rates of ALS in
men and women for Western Washington State, Harrison County,
Texas, and Ireland. The prevalence of ALS is estimated at
4-6 out of 100,000 people, and that the 60-75 year old age-group
is 33/100,000 in a population for men and 14/100,000 in a
population for women (Majoor-Krakauer, Willems, & Hofman,
2003).
In the United States there are around 20,000 to 30,000 people
living with ALS (Miller, Gelinas, & O’Connor, 2004).
It's still not really known what causes this disease, and
predicting who is at risk for it can only be done for a very
small population who are at risk because of their genes.
Women are somehow protected from the disease, because women
tend to develop it at a later age compared to men and to develop
it less. However, there is still no
known reason why this happens. A lot of research still
needs to be done.
Around 20% of people with a family history of the disease
have it because of a SOD-1
gene abnormality (Miller, Gelinas, & O'Connor, 2004).
Only one to two percent of people with ALS actually have the
gene abnormality. This causes people to lack encouragement
to be tested since there's a very high chance of having ALS
with no gene abnormality. The gene is supposed to code for
the protein SOD-1, which helps cleanse body cells of waste
products known as free
radicals (Miller, Gelinas, & O’Connor, 2004).
Since free radicals are not being cleansed from the body,
the free radicals destroy motor neurons and eventually the
motor neurons die.
Other causes of ALS are theorized, such as having excessive
stimulation of the motor nerve cells by the amino acid glutamate
(Miller, Gelinas, & O’Connor, 2004). This is an
amino acid necessary for signal relay throughout the motor
neurons, normal growth, and metabolism. It's like a messenger
between neurons. However, too much stimulation damages the
neurons. Although it's known that over-stimulation of motor
neurons causes damage, it's not completely known in most cases
why a person gets ALS.
ALS is only one of a couple motor neuron diseases. Symptoms
are shared by other diseases and conditions such as progressive
muscular atrophy, HIV,
multiple
sclerosis, bulbar and pseudobulbar palsy,
and neurofibroma
of the dorsal spinal
cord. ALS is usually diagnosed about 6-18 months after
the onset of symptoms because of diagnostic difficulties when
symptoms first begin, clinical variability, lack of specific
biological
markers, and the subclinical progression in the early
course (Calvo et al., 2009).
Symptoms may first appear by having difficulty talking, chewing
or swallowing, and/or having problems with breathing while
eating. Some people start by having difficulty walking, clumsiness,
weakness in one or both legs, and tripping. Others start with
difficulty in hand and arm movements, such as buttoning a
shirt, combing or washing their hair, or anything needing
shoulder abduction. Eventually all these symptoms will present
them self in a person with ALS; however, each person may begin
with one or many symptoms.
Doctors usually use the El
Escorial criteria to diagnose ALS, which has two panels
(Mitchell & Borasio, 2007). The first panel is from suspected
to definite, for lower and upper motor neuron signs in one
or more regions, is probable with laboratory support criteria
of lower and upper motor neuron signs in one region, or upper
motor neuron signs in one or more regions with EMG evidence
of acute denervation
in two or more limbs.
The second panel includes direct and indirect symptoms that
are attributable to ALS. Direct symptoms are due to motor
neuronal degeneration, and indirect symptoms are a result
of primary symptoms. Direct symptoms are those already mentioned,
however, indirect symptoms include psychological disturbances,
sleep disturbances, constipation, drooling, thick mucus secretion,
symptoms of chronic hypoventilation,
and pain (Mitchell & Borasio, 2007).
There are other tests that can help identify someone who
has ALS, and may help clinical studies be more objective.
The most widely used scale in clinical trials is the ALS
functional rating scale (Mitchell & Borasio, 2007).
Scales are used to assess quality of life status.
Other physical examinations have to be conducted, such as
checking for toxicity in cells with free
radicals and oxidant
stress. Blood work should be done to check and rule out
HIV and other diseases, as well as levels like creatine kinase
tested
to determine any effect on muscle functioning. Paraspinal
electromyography (EMG) is a muscle biopsy that should be done
to check for nerve
conduction. MRI
scans of cervical and lumbar regions
can also rule out other conditions like neurofibroma.
Even though ALS is not curable, it is treatable. Immunomodulatory
treatment globulin
can be done in patients with severe weakness (Mitchell &
Borasio, 2007). This helps muscle proteins develop, and to
start working more on cleaning the muscle cells of any excess
metabolic materials. Immunoglobins are also antibodies, which
are plasma proteins needed to defend the body against bacterial
and viral infections. Having a healthy immune system will
help prevent autoimmune diseases (Pyne, Ehreinstein, &
Morris, 2002).
To extend the survival of people with ALS the U.S. Food and
Drug Administration has approved riluzole
(Miller, Gelinas, & O’Connor, 2004). Also, other
sources are working to find ways to control mitochondrial
dysfunction that comes with muscle wasting in ALS. Vitamins
C and E, glutathione,
and coenzyme
Q10 can also help with the mitochondrial defense strategy
against the free oxygen radicals (Du & Yan, 2010). These
are all normal components of muscle cells, but supplementing
them to a person with ALS can help delay onset of symptoms.
Vitamin E 1,000 mg/day and riluzole has shown to cause a slower
progression of symptoms, and improvement in blood markers
of oxidative stress (Miller, Gelinas, & O’Connor,
2004).
Specific symptoms can be treated, such as muscles stiffness,
weakness, spasms, aches, and cramps, to help reduce discomforts
that come with ALS. Patients should be given aid to prevent
pressure and pain sores by having someone help with changing
positions frequently. Physical activity with the muscles that
have not been affected, and help with movement of the muscles
that have already been affected, is very important to prevent
deterioration from reasons other than ALS. Range of motion
exercises should help minimize contractures due to muscle
disuse, which will help to ease patient discomfort (Stopka
& Todorovich, 2008). Being assisted with normal living
patterns will be necessary, and adapted techniques - such
as the use of computer technology to help with communication
and eventually artificial breathing - will have to be used.
People with ALS not only need physical help, but they need
to have activities in which interactive, social, and emotional
abilities are being exercised. Patients with ALS may feel
hopeless if there's a lack in treatment and having no cure;
however, they usually have intact cognitive functioning. Having
someone to communicate with, and to keep their attention moving
and focused, can be very good. Many board games can still
be played with assistance. Joining a group of other people
who have ALS can help produce good emotions, high morale,
and to keep communication flowing with others. The use of
computer technology can also help with daily activities such
as turning T.V., lights, and phone on and off, and to help
with opening and closing of doors.
Having vigorous and intense exercise can harm a patient with
ALS because their muscles are not going to be able to handle
it. However, endurance exercise training increases the capacity
of major antioxidant enzymes, reduces oxidative stress, and
increases mitochondrial
capacity in the muscle. So, regular exercise can help patients
with ALS (Patel & Hamadeh, 2009).
Moving the muscles that are working, with small resistance
exercises every day, can help the mitochondria
keep functioning correctly. Exercises, such as rowing and
fencing, can be done even if the legs are paralyzed. Exercise
can also help protect the neurons by easing motor deficit,
and inciting the formation of new nerve cells (Patel &
Hamadeh, 2009). It has been found that regular, low, and moderate
intensity exercise delays the onset of ALS; a resistance exercise
program like pushing and pulling a block, or a bowling ball
on the floor. The exercises can vary depending on the stage
of ALS that a person is in. If they are still able to move
their legs, walking and incremental bicycling should be done.
Having a secured spot where the person with ALS can move
a bit, but not necessarily fall because of loss in muscle
coordination and strength, is best when developing an exercise
program. Low impact aerobic exercises are also key to strengthen
unaffected muscles, improving the cardiovascular system, as
well as helping the nervous system prevent depression. Equipment
can help the patient do some exercises on their own. Walkers,
braces, ramps, and wheelchairs can help the person with ALS
move and keep doing activities as their muscles degrade slowly
with the disease. Range of motion and stretching should always
be done, and toward the later stages of ALS a person should
stretch out their muscles even if they are not movable. Engaging
in those types of movements, even if they are small like toe
raises, butterflies, or neck stretches, can help maintain
function and prolong ability as long as possible. This also
prevents atrophy of the muscles because of disuse.
Unfortunately, much research is still needed to find a cure
for ALS, as currently there is no cure. However, there are
people who have lived for more than three years with ALS.
Dr.
Stephen Hawking is one of the most well known people alive
who has ALS. He uses voice synthesizers, predictive text entry
systems, and other computer technologies to still do his research
on astrophysics, and to communicate with others as well as
give conferences. Through incorporating physical activity,
a person diagnosed with ALS can have a better outlook on life.
Dr. Hawkings' survival for more than 40 years with ALS can
definitely show that there is hope for people who are diagnosed
with this condition.
references
Biography: Melissa Ortiz
was born in Colombia on May 5, 1990, where she experienced
great lessons of integrity and compassion. Her mother, Patricia
Bedoya, saw better opportunities in the United States and
they relocated there in 2000. Through hard work, and with
an everlasting dedication to self-improvement, Melissa obtained
a Bachelor of Science in Health Education & Behavior on
August 2011 from the University of Florida.
Also, at UF within the College of Nursing, she worked in
research on Promoting Mental Health among Latinos in Rural
Areas. She is a co-author in the article entitled Promotoras
in Mental Health: A Review of English, Spanish, and Portuguese
Literature published in the Journal
Family & Community Health (also PubMed.gov,
International
Nursing Library). Currently, she attends Edward Via College
of Osteopathic Medicine – Carolinas Campus - as a first
year student with aspirations of attending under-served and
disadvantaged populations.
(back
to pelinks4u homepage) |



